
— Studio collaborativo condotto da Zhejiang CDC, Macro & Micro-Test e China CDC pubblicato su Frontiers in Cellular and Infection Microbiology
Panoramica dello studio
Nel maggio 2026, Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) ha pubblicato un articolo guidato dal Centro provinciale per il controllo e la prevenzione delle malattie dello Zhejiang (Zhejiang CDC), con il team di bioinformatica della Beijing Macro & Micro-Test Bio-Tech Co., Ltd. e l'Istituto nazionale per il controllo e la prevenzione delle malattie trasmissibili (China CDC) come coautori. Lo studio è intitolato:
"Identificazione e analisi filogenetica di sette ceppi di Brucella abortus nello Zhejiang, Cina."

Questo studio rappresenta la prima analisi sistematica di tracciabilità filogenetica basata sull'intero genoma di Brucella abortus (B. abortus) nella provincia di Zhejiang, in Cina. Il team ha analizzato sette isolati raccolti tra il 2015 e il 2025 (quattro di origine umana e tre di origine bovina provenienti da Jinhua, Quzhou e Ningbo). I risultati forniscono prove genomiche sull'origine e sulle vie di trasmissione di questa "specie dominante settentrionale" in un'atipica regione epidemica meridionale della Cina orientale.
Contesto e significato
La brucellosi è una malattia zoonotica causata da batteri del genere Brucella. Brucella abortus infetta principalmente i bovini, ma può causare malattie anche nell'uomo. In Cina, la brucellosi presenta una marcata variabilità geografica: l'incidenza più elevata si riscontra nelle province settentrionali (ad esempio, Mongolia Interna, Shanxi, Heilongjiang). Al contrario, le province meridionali, tra cui lo Zhejiang, sono state storicamente dominate da Brucella melitensis, con pochissimi casi segnalati di B. abortus. Questa disparità regionale rende la caratterizzazione genetica e l'individuazione della fonte di B. abortus nello Zhejiang una priorità fondamentale per la salute pubblica.
Metodi e risultati principali
Il team di ricerca ha adottato una strategia multiforme che combina biologia molecolare e bioinformatica:
1.Identificazione e tipizzazione di base degli agenti patogeni
L'analisi mediante PCR del gene BCSP-31 e AMOS-PCR ha confermato che tutti e sette gli isolati erano B. abortus.
La tipizzazione multilocus a sequenza (MLST) basata su nove geni housekeeping ha rivelato che tutti gli isolati appartenevano al tipo di sequenza ST2, indicando un'elevata omogeneità genetica tra i ceppi di B. abortus circolanti nello Zhejiang.
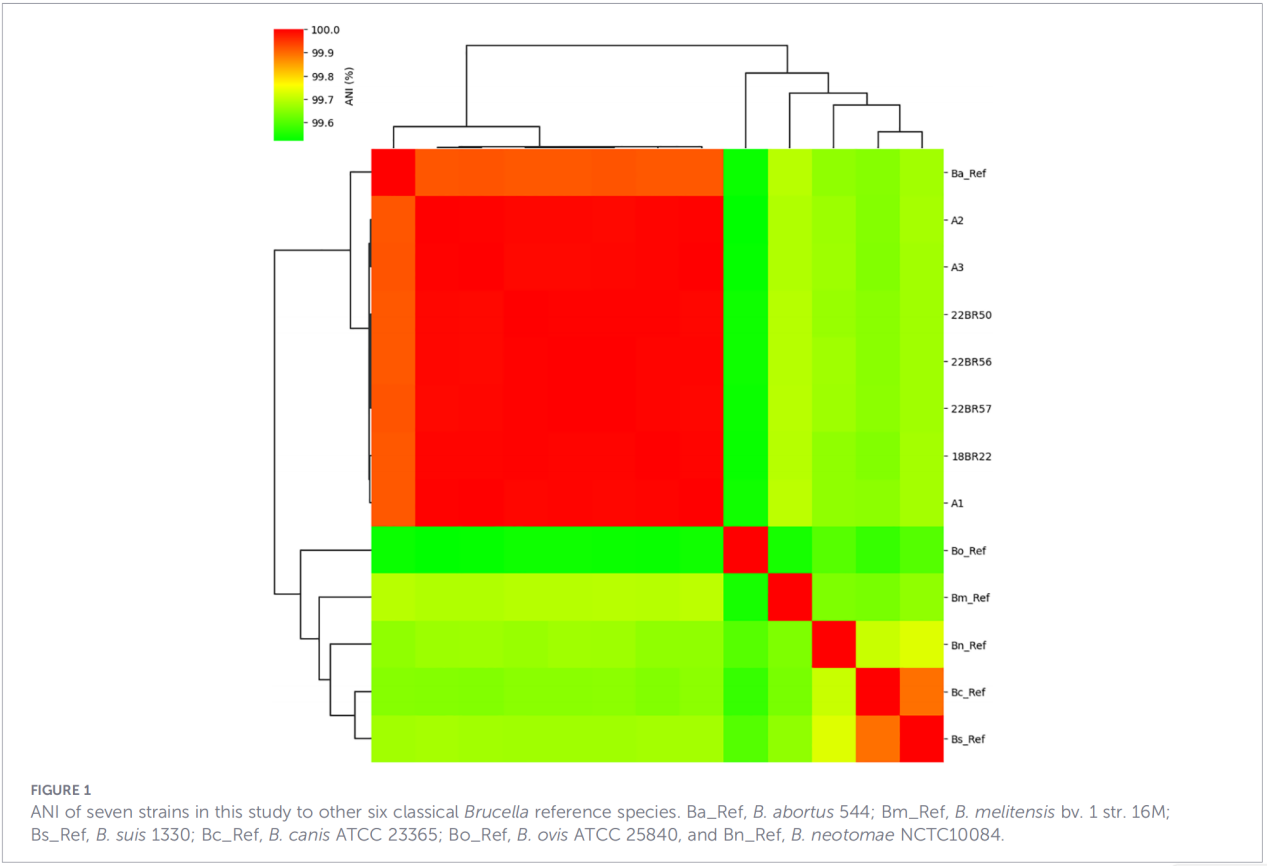
2.Caratterizzazione dell'intero genoma
Il sequenziamento dell'intero genoma è stato eseguito sulla piattaforma Illumina NovaSeq. L'analisi dell'identità nucleotidica media (ANI) ha mostrato che gli isolati dello Zhejiang condividevano fino al 99,99% di similarità con il ceppo di riferimento B. abortus 544.
L'analisi del pan-genoma ha rivelato una popolazione altamente conservata: sono stati identificati 3.084 geni core, insieme a soli 10 geni shell, e non sono stati rilevati geni soft core o cloud.
3.Profili di geni di virulenza e resistenza antimicrobica
Sono stati previsti complessivamente 68 fattori correlati alla virulenza, che comprendono vie metaboliche classiche come la biosintesi del LPS, il sistema di secrezione T4SS e il sistema di regolazione a due componenti BvrR-BvrS. In particolare, tutti gli isolati erano privi dei geni di adesione bmaA e btaF. L'analisi dei geni di resistenza ha rilevato solo il gene mprF nel database CARD, senza identificare altri determinanti di resistenza.
 4. Ricostruzione filogenetica e tracciamento della trasmissione
4. Ricostruzione filogenetica e tracciamento della trasmissione
L'analisi dei polimorfismi a singolo nucleotide del genoma centrale (cgSNP) ha collocato gli isolati dello Zhejiang in una posizione specifica nell'albero filogenetico globale. I risultati hanno mostrato che i ceppi dello Zhejiang formano un gruppo monofiletico insieme a ceppi provenienti da Russia, Mongolia e diverse province della Cina settentrionale (Ningxia, Heilongjiang, Mongolia Interna, Hebei, Gansu, Pechino). Questo gruppo si suddivide ulteriormente in tre sottocladi distinti (Clade 1-3), suggerendo molteplici eventi di introduzione indipendenti.
Conclusioni e implicazioni
Questo studio fornisce il primo set di dati genomici ad alta precisione di B. abortus nella provincia di Zhejiang e porta a diverse conclusioni chiave:
- Cristoar patrimonio genetico– I ceppi di B. abortus che circolano nello Zhejiang appartengono al tipo ST2, sono altamente conservati a livello genomico e rappresentano un tipico lignaggio della brucellosi bovina.
2. Eviincidenza della trasmissione interregionale– L'analisi filogenetica non supporta l'esistenza di un lignaggio endemico indipendente nello Zhejiang. Al contrario, i dati suggeriscono fortemente che questi ceppi abbiano avuto origine nella Cina settentrionale e possano condividere un background evolutivo comune con ceppi provenienti da Russia e Mongolia. La presenza di tre sottocladi implica molteplici eventi di introduzione separati.
3. Implicazioni per la salute pubblica– I risultati sottolineano l'importanza della sorveglianza genomica per la brucellosi anche in regioni tradizionalmente non endemiche come lo Zhejiang. Sebbene il numero attuale di casi sia basso, strumenti ad alta risoluzione come il cgSNP possono tracciare efficacemente la fonte dei focolai importati e fornire prove scientifiche per interrompere le catene di trasmissione associate al trasporto interprovinciale di bestiame.
Questo lavoro non solo colma una lacuna nella ricerca nella provincia di Zhejiang, ma fornisce anche nuovi dati di base per la sorveglianza degli agenti patogeni e la valutazione del rischio di brucellosi nella regione del delta del fiume Yangtze.
Informazioni sul documento:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identificazione e analisi filogenetica di sette ceppi di Brucella abortus nello Zhejiang, Cina. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Data di pubblicazione: 10 giugno 2026